Background
In this section we describe the methods available to access and
extract the gated data within a GatingSet. It is important
to note here that the gates essentially act as filter
for the data and may be utilize to subset the original data.
library(flowWorkspace)
library(flowCore)
library(CytoverseBioc2023)## Warning: replacing previous import 'flowViz::contour' by 'graphics::contour'
## when loading 'flowStats'Gating paths: get and set
Let’s first look at the gates that are attached to the
GatingSet. For this example, we will make use of a
GatingSet that we have previously prepared.
gs <- make_transformed_gs(add_gates = TRUE)
gs## A GatingSet with 4 samplesWe have previously used plot(gs) to visualize the gating
hierarchy as a tree as shown below:
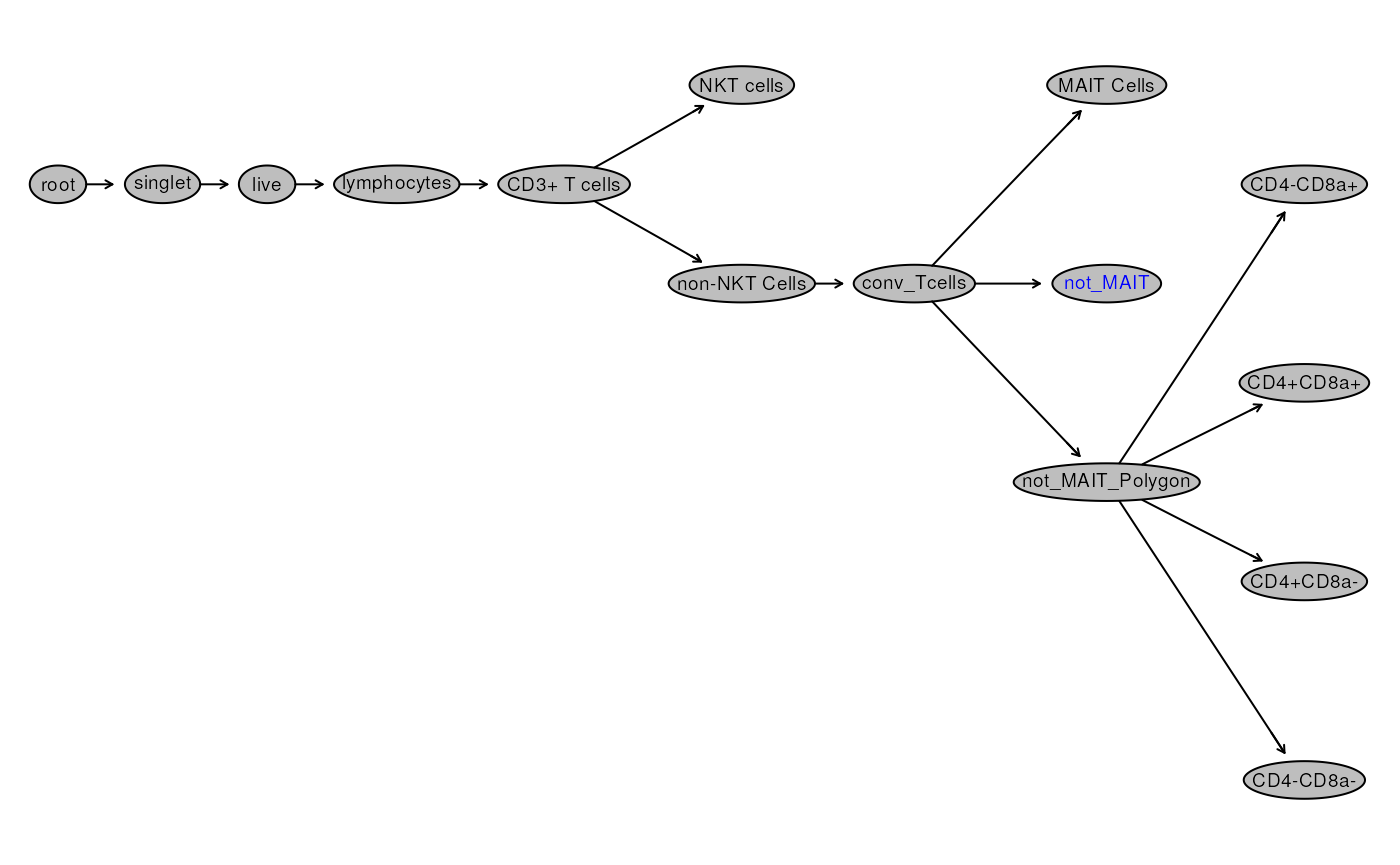
To extract the full gating path we can use
gs_get_pop_paths method.
# get all paths
gs_get_pop_paths(gs)## [1] "root"
## [2] "/singlet"
## [3] "/singlet/live"
## [4] "/singlet/live/lymphocytes"
## [5] "/singlet/live/lymphocytes/CD3+ T cells"
## [6] "/singlet/live/lymphocytes/CD3+ T cells/NKT cells"
## [7] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells"
## [8] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells/conv_Tcells"
## [9] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells/conv_Tcells/MAIT Cells"
## [10] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells/conv_Tcells/not_MAIT"
## [11] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells/conv_Tcells/not_MAIT_Polygon"
## [12] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells/conv_Tcells/not_MAIT_Polygon/CD4-CD8a+"
## [13] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells/conv_Tcells/not_MAIT_Polygon/CD4+CD8a+"
## [14] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells/conv_Tcells/not_MAIT_Polygon/CD4+CD8a-"
## [15] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells/conv_Tcells/not_MAIT_Polygon/CD4-CD8a-"When there are a lot of gates, this list can become very long as each
node is shown as a distinct path. Suppose you would like to only view
the leaf nodes. For this, we use gs_get_leaf_nodes.
# get leaf nodes
gs_get_leaf_nodes(
gs,
ancestor = "root") # can select leafs of a specific node## [1] "/singlet/live/lymphocytes/CD3+ T cells/NKT cells"
## [2] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells/conv_Tcells/MAIT Cells"
## [3] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells/conv_Tcells/not_MAIT"
## [4] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells/conv_Tcells/not_MAIT_Polygon/CD4-CD8a+"
## [5] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells/conv_Tcells/not_MAIT_Polygon/CD4+CD8a+"
## [6] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells/conv_Tcells/not_MAIT_Polygon/CD4+CD8a-"
## [7] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells/conv_Tcells/not_MAIT_Polygon/CD4-CD8a-"To change the name of a node, for instance,
CD4-CD8a- above is not super clear and we want to
update this to Double Negative CD3+ T cells for
readability, we can use gs_pop_set_name method.
# rename node
gs_pop_set_name(
gs,
"CD4-CD8a-",
"Double Negative CD3+ T cells")## $`4000_TNK-CR1`
## NULL
##
## $`4001_TNK-CR1`
## NULL
##
## $`4002_TNK-CR1`
## NULL
##
## $`4003_TNK-CR1`
## NULL
# check
gs_get_leaf_nodes(gs)## [1] "/singlet/live/lymphocytes/CD3+ T cells/NKT cells"
## [2] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells/conv_Tcells/MAIT Cells"
## [3] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells/conv_Tcells/not_MAIT"
## [4] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells/conv_Tcells/not_MAIT_Polygon/CD4-CD8a+"
## [5] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells/conv_Tcells/not_MAIT_Polygon/CD4+CD8a+"
## [6] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells/conv_Tcells/not_MAIT_Polygon/CD4+CD8a-"
## [7] "/singlet/live/lymphocytes/CD3+ T cells/non-NKT Cells/conv_Tcells/not_MAIT_Polygon/Double Negative CD3+ T cells"Exercise
- Run
gs_get_pop_paths(gs)by include apath = 1argument. How is the output different compared to not providingpath? What happens when you change this topath = 2? - Suppose you do not know what is the parent population for
Double Negative CD3+ T cells, what is an easy approach to get this information? Hint: Tryhelp(gs_pop_get_paths)to check other available methods.
Extracting filtered data
Often, users may also want to extract the embedded expression data
for a specified gated population for some downstream application. The
cytoverse makes it super easy to extract this data.
Moreover, the extracted data is preserved as a cytoframe or
a cytoset for ease of use.
The 2 main methods are: 1. gs_pop_get_data 2.
gh_pop_get_data
# extract data
extracted <- gs_pop_get_data(gs,
y = "live",
inverse.transform = FALSE)
extracted## A cytoset with 4 samples.
##
## column names:
## FSC-A, FSC-H, SSC-A, B515-A, B610-A, B660-A, B710-A, B780-A, G575-A, G610-A, G660-A, G710-A, G780-A, R670-A, R730-A, R780-A, U390-A, U450-A, U500-A, U570-A, U660-A, U740-A, U785-A, V450-A, V510-A, V570-A, V605-A, V655-A, V710-A, V750-A, V785-A, remove_from_FS_FM, Time
##
## cytoset has been subsetted and can be realized through 'realize_view()'.Exercise
- Try running the above code as
gh_pop_get_data. How are the results different? - Extract the data from the gate
Double Negative CD3+ T cellsfor the 1st sample and store it assample_1. Transform the expression value forFSC-A. What happens to the data ings?Hint
# plot
ggcyto(gs,
subset = "Double Negative CD3+ T cells",
aes(x = "FSC-A", y = "SSC-A"))+
geom_hex(bins = 256)GatingSet. This is because we did not create a new copy of
the data using realize_view method. Importantly, this
further highlights that the cytoframe,
cytoset, and the GatingSet are all pointing to
the same data!
- what does
inverse.transformargument do? - Run the following code:
This is only for example.
another_CD3_gate <- matrix(c(-Inf,Inf), nrow = 2, ncol=1)
colnames(another_CD3_gate) <- "FSC-A"
another_CD3_gate <- rectangleGate(.gate = another_CD3_gate, filterId = "CD3+ T cells")
gs_pop_add(gs, parent = "non-NKT Cells", gate = another_CD3_gate)
recompute(gs)
plot(gs)Now try to extract the CD3+ T cells population using
either gs_ or gh_ methods as above.
Saving extracted data
Depending on the method called, the extracted data is either
preserved as a cytoframe or a cytoset. As you
experienced in the exercise, manipulating the extracted without
performing a deep copy using realize_view leads to
alteration of the data within the GatingSet object. As
such, if extracting for further downstream application, it maybe
worthwhile to save the data. For this, please refer to the Importing and Basics of working with
FCS files for more details.
Conclusion
In this brief section we demonstrated how users/analysts could
extract the embedded data from a GatingSet. Feel free to
explore the flowWorkspace vignette for additional available
methods. In the next section, Reporting, we will go over how to
extract various statistics regarding the gated populations.